WHAT IS THALASSEMIA
History of Thalassemia:

Thalassemia was first named after Dr. Cooley. It wasn’t until 1946 that the underlying causes of the disease were better understood. The formal treatment of thalassemia began in the 1960s, when blood transfusion therapy was introduced for the first
identified in regions associated with Greece, Italy, Cyprus, and the Mediterranean basin. In 1925, two physicians from Michigan, USA — Dr. Cooley and Dr. Lee — diagnosed this blood disorder while conducting research on anemia. Initially, the condition was referred to as Cooley’s Anemia,
time to manage the condition in affected children. Since then, this has remained the primary method of treatment,aiming to prolong and improve the quality of life of children diagnosed with thalassemia.
Although thalassemia is found in many parts of the world, it is more prevalent among people of Greek, Italian, Cypriot, Middle Eastern, South Asian, and African descent.
Causes and Risk Factors:
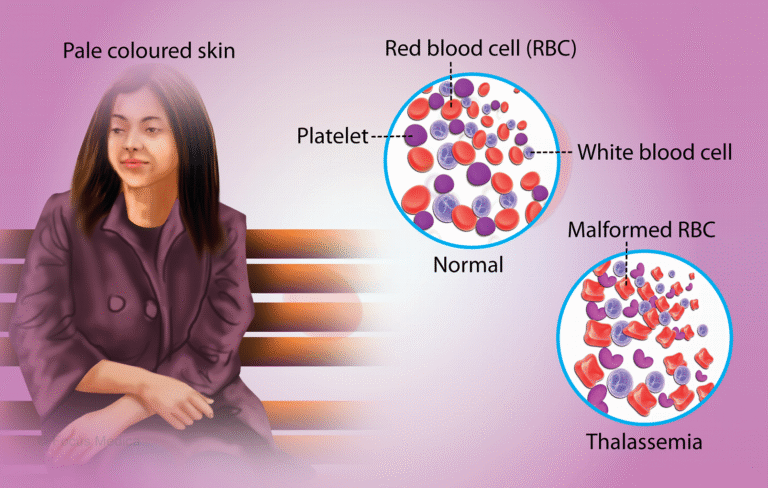
Thalassemia is a genetic blood disorder that affects the body’s ability to produce hemoglobin. When both parents are carriers (Thalassemia Minor), there is a significant risk that their child will be born with Thalassemia Major, the more severe form of the disease. It is an inherited condition passed from parents to children through genes. In individuals affected by thalassemia, the body’s natural process of producing healthy red blood cells is impaired, resulting in chronic anemia. As a result, patients require regular blood transfusions—often on a monthly basis—for survival. Their entire life becomes dependent on this ongoing treatment.
Consanguineous (within-family) marriages significantly increase the risk of thalassemia transmission, contributing to a higher prevalence of the disease in certain communities.
Types of Thalassemia:
From a genetic standpoint, thalassemia is broadly classified into two main types: Alpha Thalassemia and Beta Thalassemia. In Pakistan, Beta Thalassemia is more prevalent, whereas Alpha Thalassemia is more commonly observed in Far Eastern countries.
Among the clinical forms, the mildest variant is known as Thalassemia Minor, while the most severe form is called Thalassemia Major. An intermediate form, with moderate severity, is referred to as Thalassemia Intermedia.
It is important to note that one type of thalassemia can never convert into another—for instance, Alpha Thalassemia cannot turn into Beta Thalassemia, and vice versa. Similarly, a Thalassemia Minor patient can never develop into a Major, nor can a Major revert to Minor. Each form is genetically fixed and determined at birth.
Thalassemia Minor:
Thalassemia Minor is not a disease, and individuals with this condition typically experience no symptoms, discomfort, or health-related complaints. It has little to no impact on a person’s daily life or life expectancy. Because it is asymptomatic, its presence can only be confirmed through laboratory testing, particularly through a specialized blood test called Hb Electrophoresis.
People with Thalassemia Minor lead completely normal lives—physically, mentally, and sexually. They are often unaware of the genetic mutation they carry, yet they can pass on thalassemia to their children.
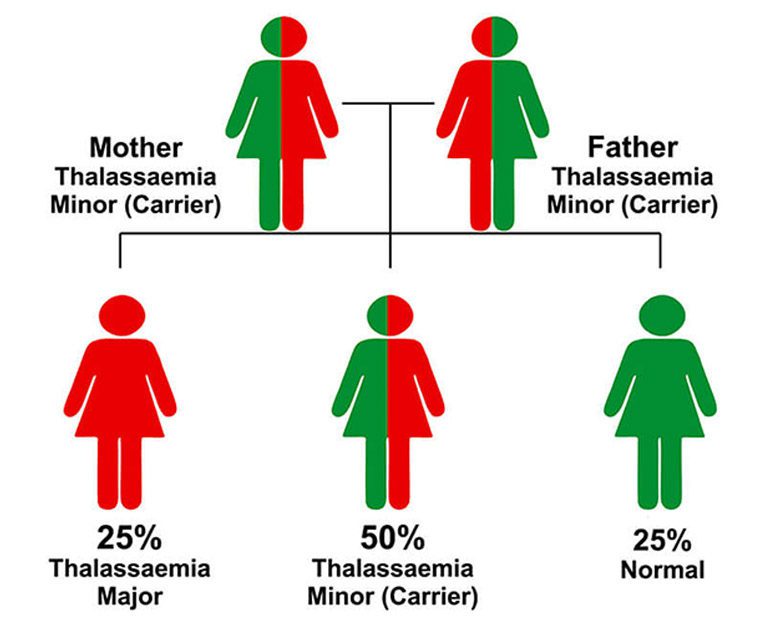
Importantly, when both parents are carriers of Thalassemia Minor, there is a high probability that their child may be born with Thalassemia Major, a far more severe and life-altering form of the disorder. While Thalassemia Minor does not manifest any clinical symptoms, its genetic transmission potential makes screening and awareness critically important, especially before marriage.
Among the clinical forms, the mildest variant is known as Thalassemia Minor, while the most severe form is called Thalassemia Major. An intermediate form, with moderate severity, is referred to as Thalassemia Intermedia.
It is important to note that one type of thalassemia can never convert into another—for instance, Alpha Thalassemia cannot turn into Beta Thalassemia, and vice versa. Similarly, a Thalassemia Minor patient can never develop into a Major, nor can a Major revert to Minor. Each form is genetically fixed and determined at birth.
Thalassemia Major:
In Thalassemia Major, the body’s ability to produce red blood cells is severely impaired, leading to chronic and life-threatening anemia. As a result, children affected by this condition require lifelong blood transfusions, often every 2 to 4 weeks, to survive. These children begin showing signs of severe anemia just a few months after birth, and their survival becomes entirely dependent on blood banks.
Thalassemia Major is a genetic and hereditary disorder that is equally passed on by both parents. When both parents are carriers of Thalassemia Minor, there is a 25% chance in every pregnancy that the child will be born with Thalassemia Major.
In this condition, the body fails to produce enough red blood cells, leading to extreme deficiency of hemoglobin. The symptoms typically begin to appear when the child is around four months old. Children with Thalassemia Major require not only frequent blood transfusions but also lifelong medication to manage iron overload, which results from repeated transfusions.
This is a serious and lifelong medical condition, and its only reliable prevention is carrier screening and awareness before marriage.
Symptoms of Thalassemia Major:
The symptoms of Thalassemia Major usually begin to appear within the first few days or months after birth. The most common and noticeable signs include:
- Yellowing of the skin (jaundice)
- Persistent weakness and fatigue
- Difficulty in breathing
- Abdominal swelling or bloating
- Recurrent cough or diarrhea, often triggered by general physical weakness
These symptoms often worsen as the child grows, making early diagnosis and intervention critical.
People with Thalassemia Minor lead completely normal lives—physically, mentally, and sexually. They are often unaware of the genetic mutation they carry, yet they can pass on thalassemia to their children.
Importantly, when both parents are carriers of Thalassemia Minor, there is a high probability that their child may be born with Thalassemia Major, a far more severe and life-altering form of the disorder. While Thalassemia Minor does not manifest any clinical symptoms, its genetic transmission potential makes screening and awareness critically important, especially before marriage.
Among the clinical forms, the mildest variant is known as Thalassemia Minor, while the most severe form is called Thalassemia Major. An intermediate form, with moderate severity, is referred to as Thalassemia Intermedia.
It is important to note that one type of thalassemia can never convert into another—for instance, Alpha Thalassemia cannot turn into Beta Thalassemia, and vice versa. Similarly, a Thalassemia Minor patient can never develop into a Major, nor can a Major revert to Minor. Each form is genetically fixed and determined at birth.
Prevalence and Statistics:
Thalassemia is becoming an increasingly serious health concern in Pakistan. Unfortunately, there has not yet been any official nationwide survey, and due to the absence of a national database, the exact number of affected individuals remains unknown.
However, according to various non-governmental and independent surveys, it is estimated that the number of children living with Thalassemia Major in Pakistan has exceeded 80,000, and approximately 6,000 new cases are diagnosed each year.
The reality is that screening for Thalassemia Minor is simple and does not require advanced equipment. It can be carried out at any hospital, basic health unit, rural health center, or dispensary.
At Children’s Hospital in Gulshan-e-Iqbal, Karachi, for instance, 12 to 14 new Thalassemia patients are reported daily, amounting to 1,200 to 1,400 new cases annually. These figures underscore the alarming growth in the number of children being born with Thalassemia each year, and highlight the urgent need for preventive screening and public health intervention.